Characterizing cancer-risk mutations (2015-2017)
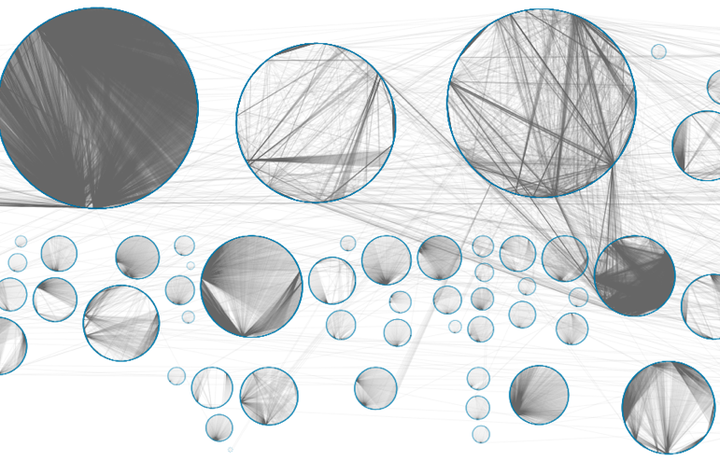 An eQTL network corresponding to the skin tissue.
An eQTL network corresponding to the skin tissue.
Genome-wide associations studies (GWAS) have identified many non-coding germline single nucleotide polymorphisms (SNPs) that are associated with an increased risk of developing cancer. However, how these SNPs affect cancer risk is still largely unknown. We used a systems biology approach to analyze the regulatory role of cancer-risk SNPs in thirteen tissues. Using data from the Genotype-Tissue Expression (GTEx) project, we performed an expression quantitative trait locus (eQTL) analysis. We represented both significant cis- and trans-eQTLs as edges in tissue-specific eQTL bipartite networks. Each tissue-specific eQTL network is organized into communities that group sets of SNPs and functionally- related genes. When mapping cancer-risk SNPs to these networks, we find that, in each tissue, these SNPs are significantly over-represented in communities enriched for immune response processes as well as tissue-specific functions. Moreover, cancer-risk SNPs are more likely to be “cores” of their communities, influencing the expression of many genes within the same biological processes. Finally, cancer-risk SNPs preferentially target oncogenes and tumor-suppressor genes, suggesting they may alter the expression of these key cancer genes. This approach provides not only a new way of understanding genetic effects on cancer risk, but also a biological context for interpreting the results of GWAS cancer studies.
Fundings
This postdoc was funded by a grant from the National Cancer Institute National Cancer Institute (1R35CA197449).